Anyoli Taly1,2, Xenón Serrano–Martín2,3,Jorge Núñez2, Carlos Chinea1*
1) Laboratorio de Polímeros, Escuela de Química, Universidad Central de Venezuela (UCV). Caracas, Venezuela. Correo electrónico: carlos.chinea@ciens.ucv.ve
2) Laboratorio de Biología y Quimioterapia de Parásitos Tropicales (ByQPAT), Instituto de Estudios Avanzados (IDEA). Caracas, Venezuela. Correo electrónico: xenonserrano@gmail.com
3) Centro de Química Orgánica, Facultad de Ciencias, Universidad Central de Venezuela (UCV). Caracas,Venezuela.
Recibido: Julio 2019; Aceptado: Agosto 2019
Texto completo (pdf)
Cita (APA)
Taly, A., Serrano-Martín, X., Núñez, J., Chinea, C. (2019). Síntesis de polímeros biodegradables de poli (D, L–lactida y L–Lactida) para emplearlos como dispositivos de liberación controlada de ciclopirox olamina, sobre la viabilidad de Leishmania Braziliensis. Revista Iberoamericana de Polímeros, 20(5), 207–220.
RESUMEN
En el presente estudio, demostramos el efecto biológico de ciclopirox olamina (CX) dispensado desde microcápsulas de poli(D, L y L–lactida), sobre el parásito trypanosomatideo Leishmania braziliensis. El mismo, es el agente causal de Leishmaniasis Cutánea, manifestación clínica preponderante de esta parasitosis en Venezuela. Los polímeros fueron preparados por el método de polimerización por apertura de anillo (ROP). En la elaboración de las microcápsulas, se empleó el método de emulsión simple para estudiar los cambios morfológicos de las microcápsulas por degradación hidrolítica y el método de doble emulsión por evaporación de disolvente para encapsular el fármaco CX. Observamos entonces que las microcápsulas de Poli(D, L–Lactida), se degradaron más rápido que las de Poli(L–Lactida), con una eficiencia de encapsulación del fármaco de 18 y 26%, respectivamente. Seguidamente determinamos que el perfil de liberación del fármaco fue bifásico, para ambos tipos de dispositivos. Finalmente, se procedió a evaluar el efecto biológico del fármaco en su forma libre y el dispensado desde las microcápsulas, sobre la viabilidad de parásitos L. braziliensis. Este último ensayo, mostró que los dispositivos liberaron controladamente el medicamento, garantizando una presión selectiva constante y con ello un efecto parasiticida sostenido.
ABSTRACT
In the present study, we demonstrate the biological effect of cyclopirox olamine (CX) dispensed from poly(D, L and L–lactide) microcapsules, on the trypanosomatid parasite Leishmania braziliensis. It is the causative agent of cutaneous leishmaniasis, a preponderant clinical manifestation of this parasitosis in Venezuela. The polymers were prepared by the ring–opening polymerization method (ROP). In elaboration of microcapsules, simple emulsion method was used to study the morphological changes of microcapsules by hydrolytic degradation and double emulsion method by solvent evaporation to encapsulate CX. We observed that microcapsules of Poly(D, L–Lactide), degraded faster than those of Poli L–Lactide, with a drug encapsulation efficiency of 18 and 26%, respectively. Next, we determined that drug release profile was biphasic, for both types of devices. Finally, we proceeded to evaluate the biological effect of drug in its free form and that dispensed from the microcapsules, on the viability of parasites L. braziliensis. This last test showed that the devices controlled release of the drug, guaranteeing constant selective pressure and thus a sustained parasiticidal effect .
INTRODUCCIÓN
Durante más de dos décadas el suministro de agentes bio–activos, que cumplen funciones en el cuerpo para promover la buena salud, extraídos de plantas y ciertos alimentos, se han ido mejorado con el uso de materiales poliméricos biodegradables, que han traído considerable atención a los investigadores en toda la comunidad científica tales como químicos de polímeros, ingenieros químicos, farmacéuticos y entomólogos, que tratan de diseñar sistemas de liberación controlada de agentes bioactivos que van de insulina a rodenticidas [1].
Debido a que la degradación de estos polímeros es por hidrólisis, existe una tendencia en la tecnología de administración de fármacos orientada hacia el uso de materiales biodegradables que no requieren la extirpación quirúrgica una vez que el suministro de medicamentos se agota, evitando costo y trauma al paciente [1], ya que el monómero de partida, producto de la degradación hidrolítica, es biocompatible y desechado por vías metabólicas [2].
Uno de los materiales más utilizados es el poliácido láctico, el cual cuenta con amplias aplicaciones en campos biomédicos, incluyendo la sutura, material de fijación de hueso, microesferas de administración de fármacos e ingeniería de tejidos [2]. El poliácido láctico (PLA) es un polímero biodegradable que se ha estudiado extensamente durante los últimos 15 años, el cual se obtiene a partir de la polimerización del ácido láctico [3] o el dímero cíclico lactida. El PLA puede ser sintetizado por dos mecanismos de reacción: polimerización por apertura de anillo (ROP, por siglas en idioma inglés) del intermediario lactida o por policondensación. Hoy en día, ROP es el método más usado, implementado en una larga escala de producción por la compañía Cargill Dow LLC en los Estados Unidos de América. Este es un proceso efectivo y complejo que requiere de pasos rigurosos de purificación de la lactida. El método más reciente permite obtener PLA a partir del monómero ácido láctico. Sin embargo, para producir polímeros de alto peso molecular es necesario asegurarse de que el agua formada sea removida [4]. El método ROP es más apropiado, a pesar de su costo, y hace que la formación del polímero sea más factible, ya que se evita la formación de agua que impide que el producto tenga un alto peso molecular.
Este polímero ha mostrado gran versatilidad como agente de liberación de fármaco, lo que favorece su uso en el tratamiento de enfermedades que requieren de métodos terapéuticos que son incómodos para el paciente, de alta toxicidad y a veces inefectivos sobre la zona afectada. En el caso de enfermedades tropicales como la leishmaniasis (la cual fue de interés para este trabajo), es un recurso factible implementar un polímero biodegradable con liberación controlada del medicamento para que el tratamiento sea más específico sobre las lesiones y genere menos traumas médicos, asegurando también una dosificación continua.
La leishmaniasis es una enfermedad parasitaria, catalogada como endémica en países como Venezuela. Posee tres manifestaciones clínicas, las cuales son leishmaniasis cutánea, mucococutánea y visceral; cada una de ellas producidas por una especie particular del género Leishmania, siendo L. braziliensis el causante principal de la leishmaniasis cutánea, la cual representa la manifestación más frecuente en Venezuela. Actualmente el medicamento empleado en el tratamiento de esta enfermedad es el Glucantime®, el cual produce efectos secundarios por su toxicidad, y traumas médicos por las constantes inyecciones que son dolorosas. Debido a las desventajas que posee este tratamiento, se plantea desarrollar sistemas biotecnológicos tales como dispositivos de liberación controlada que permitan suministrar un fármaco a bajas concentraciones, de manera localizada y sostenida, mejorando su efecto terapéutico de tal manera que generen un bienestar en el paciente. En el diseño de los dispositivos de liberación controlada, se integra el medicamento en el material polimérico, generando microcápsulas que permiten liberar el contenido de fármaco de manera sostenida, por medio de una degradación hidrolítica. El medicamento en estudio fue ciclopiro xolamina, el cual ha mostrado efecto parasiticida sobre L. braziliensis, lo que permite estudiar nuevos prospectos de fármacos para una futura aplicación sobre esta enfermedad.
PARTE EXPERIMENTAL
Síntesis de poli(D, L–lactida) y poli(L– lactida) por apertura de anillo en masa. En ampollas de vidrio (bajo atmósfera inerte) se añadieron los monómeros (DL–lactida y L–lactida), disueltos en diclorometano, seguido de alícuotas de una suspensión de cloruro de estaño (II) dihidratado en diclorometano, y de una solución de 1–dodecanol en diclorometano; las cuales representaron un 0,2 y 0,05% de la masa total de los monómeros, respectivamente. Posteriormente, la mezcla fue homogeneizada y se procedió a retirar todo el solvente haciendo vacío; seguidamente se sellaron las ampollas y se sumergieron en un baño térmico a 160ºC durante 8 horas. Una vez culminado el proceso de síntesis, se guardaron las ampollas en el refrigerador para detener la reacción. Los polímeros, poli (DL–lactida) (PDL) y poli (L–lactida) (PL), fueron recolectados, purificados, y almacenados en atmósfera inerte para su posterior caracterización por Resonancia Magnética Nuclear (1H–RMN y 13C–RMN).
Microencapsulación de los polímeros (sin contendido de CX) por el método de emulsión simple por evaporación de disolvente. Una solución acuosa de 25 mL de polivilalcohol (PVOH) al 1% p/v se colocó en un reactor con agitación mecánica a 700 rpm, luego, se añadió una solución entre 70–100 mg de polímero en 6 mL de diclorometano. Se mantuvo la agitación a esa velocidad durante 30 minutos con las soluciones aun en frío, al culminar este tiempo, se continuó agitando cuatro horas y media a temperatura ambiente. Una vez finalizado el proceso, se recolectaron las microcápsulas por centrifugación, se lavaron cuidadosamente con agua destilada tibia, se secaron al vacío y se guardaron en atmósfera inerte para la realización de ensayos in vitro.
Estudio de la degradación de las microcápsulas sin contenido de CX. Ensayo in vitro en condiciones fisiológicas humanas. Se tomaron entre 3–5 mg de cada microcápsulas vacías y se añadieron a 1 mL de solución buffer de fosfato a pH = 7 dispuestos en tubos de ensayo, los cuales fueron sumergidos en un baño termostático a una temperatura constante de 37°C durante un período de tiempo que permitiera estudiar la degradación de las microcápsulas, hasta que perdieran su morfología esférica. Para estudiar estos cambios en la morfología de los dispositivos, se tomaron alícuotas cada cierto tiempo y se observaron en un microscopio óptico.
Microencapsulación de los polímeros cargados con CX por el método de doble emulsión por evaporación de solvente. Se prepararon dos formulaciones de los polímeros obtenidos, PL y PDL. Para preparar la primera emulsión, se dispuso en un embudo de adición 1 mL de una solución de agar–agar al 0,2% p/v donde había disuelto 8 mg de cliclopirox olamina. Esta fase fue añadida gota a gota sobre 6 mL de una solución polimérica en diclometano al 1,7% p/v la cual se encontraba en agitación ultrasónica y a una temperatura entre 12–15°C. Una vez culminada la adición, se continuó la agitación durante 10 minutos aproximadamente. Posteriormente, esta primera emulsión se añadió a 25 mL de una solución acuosa de PVA al 1% p/v la cual estaba en agitación mecánica a 800 rpm y en un baño de hielo a 4–6°C. Durante 30 minutos se mantuvo la agitación y la temperatura anterior; luego se retiró el baño de hielo y se dejó agitando la doble emulsión a la misma velocidad durante 4 horas y media a temperatura ambiente.
Una vez culminado el proceso de encapsulación, se recolectaron las microcápsulas por centrifugación y se lavaron con agua tibia para retirar el exceso de PVA y agar–agar. Posteriormente fueron secadas al vacío y guardadas en atmósfera inerte para su posterior caracterización por MEB y realización de ensayos in vitro.
Eficiencia de encapsulación de CX en las microcápsulas. Para determinar la cantidad de CX contenido en las microcápsulas de cada formulación, se procedió a disolver entre 4–6 mg de las mismas en 10 mL diclorometano por duplicado, para asegurar la liberación total del medicamento en la solución. Luego la concentración del fármaco en estas soluciones fue determinada por espectrofotometría UV–Visible a una longitud de onda de 306 nm.
La eficiencia de encapsulación del fármaco (E.E) se expresó como una relación entre la masa de fármaco encapsulada y la masa de fármaco inicial, y además se expresó también como rendimiento de producción de cápsulas (R, masa neta de microcápsulas entre la masa total de polímero + droga usada en formulación), y contenido de droga en las microcápsulas (C.D, masa de fármaco encapsulada entre masa de microcápsulas) [5,6].
Perfil de liberación de CX por degradación de las microcápsulas. Ensayo in vitro en medio LIT. Con la finalidad de realizar el perfil de liberación mencionado, fueron dispuestas 3 fiolas con 5 mL de medio LIT, a las cuales se añadieron las siguientes condiciones, respectivamente: a) 1,65 mg de microcápsulas de PDL cargadas con Cx; b) 2,8 mg microcápsulas de PL cargadas con CX, y c) 1 mg de microcápsulas de PL sin fármaco. Seguidamente, cada condición fue incubada a 29°C, en agitación constante, por un espacio de 4 horas. Transcurrido este período de tiempo, se procedió a tomar alícuotas de cada fiola (medidas por duplicados), las cuales fueron dispuestas en una placa de 96 pozos. Seguidamente se realizaron mediciones a una longitud de onda de 306 nm, durante 8 días continuos.
Determinación de la viabilidad de promastigotes de Leishmania braziliensis en presencia de microcápsulas cargadas con CX mediante conteos directos. Curva de crecimiento. Con la finalidad de realizar esta determinación, fueron dispuestas 18 fiolas, a las cuales se les agregó 1·106 parásitos/mL, en 5 mL de medio LIT. Se realizaron series por triplicado. Las condiciones añadidas bajo esterilidad fueron las siguientes: Condición A: parásitos sin presión selectiva; condición B: parásitos expuestos a 30 µM CX, (concentración del EC50 determinado por Pesquera [5]); condición C: parásitos expuestos a 1,68 mg de PDL con CX; condición D: parásitos expuestos a 2,8 mg de microcápsulas de PL cargadas con CX (cuya concentración teórica liberada por ambos dispositivos, de acuerdo a estos pesos, es 30 µM); condición E: parásitos expuestos a 1 mg de microcápsulas de PL sin CX; condición G: parásitos expuestos a 1 mg de microcápsulas de PDL sin CX. Cada condición, se mantuvo a 29°C, tomándose alícuotas de todas las condiciones cada 24 horas para efectuar conteos directos de proliferación parasitaria en la cámara de Neubauer por un total de 8 días continuos.
RESULTADOS Y DISCUSIÓN
Síntesis de poli(D, L–lactida) y poli(L–lactida) por apertura de anillo en masa. Poli (D,L–lactida) (PDL): Se obtuvo un polímero sólido de color blanco brillante, de aspecto espumoso. Rendimiento: 0,1450 g; 14,0%. 1H–RMN (CDCl3) δ (ppm): 1,57 (m, 3H); 5,17 (m, H). 13C–RMN (CDCl3) δ (ppm): 16,7; 69,09; 169,38.
Poli (L–lactida) (PL): Se obtuvo un polímero sólido color blanco, de aspecto cristalino. Rendimiento: 0,8070 g; 58,7%. 1H–RMN (CDCl3) δ (ppm): 1,56 (d, 3H); 5,16 (q, H). 13C–RMN (CDCl3) δ (ppm): 16,66; 69,02; 169,61.
Del espectro 13C–RMN para PL se observó una única señal en la región de 170 ppm (correspondiente al carbono carbonílico), que nos permitió decir que la estereosecuencia microestructural del polímero es isotáctica. En el caso de PDL, se observó estereosecuencias de hexadas en la región comprendida entre (169,8–169 ppm) reportadas por Bero y col. [7],donde se encuentran tres bandas anchas producto del solapamiento de la señal de carbono carbonílico. En la banda que se encuentra entre 169,5–169,8 ppm incluye las hexadas siiis, iiiis, iiiii y siiii; la segunda banda (169,3–1969,5 ppm) incluye las hexadas iiisi, isiii, iisii, sisii, iisis y sisis y la tercera banda (169,1–169,2 ppm) incluye las hexadas isisi. Estos desdoblamientos de las señales indican que el polímero presenta una estéreosecuencia aleatoria (polímero heterotáctico).
Estas tacticidades confieren un arreglo más ordenado en el caso de PL (isotáctico), que lo hace semicristalino, mientras que PDL por ser heterotáctico posee un arreglo amorfo; lo que produce una diferencia, significativa, en la degradación hidrolítica de estos polímeros en un medio acuoso.
Estudio de la degradación de las microcápsulas sin contenido de CX. Ensayo in vitro en condiciones fisiológicas humanas. Los cambios morfológicos de las microcápsulas de PL y PDL fueron evaluados por la degradación hidrolítica en una solución de PBS a pH = 7 a 37°C, en función del tiempo. Es importante mencionar, que aunque las condiciones fueron semejantes a las del cuerpo humano, no significa que el comportamiento evaluado en este ensayo in vitro corresponda al que se observaría en el medio fisiológico, debido a que en sistemas in vivo hay factores, además del pH y temperatura, que pueden influir fuertemente en la degradación hidrolítica de estos polímeros biodegradables, como las enzimas (algunas hidrolasas en el cuerpo humano, que tienen preferencia por hidrolizar las zonas amorfas de los polímeros biodegradables [8]). La intención de este ensayo, fue ver cómo la degradación de un polímero amorfo (PDL) y semi–cristalino (PL) de polilactida, afecta el cambio morfológico de las microcápsulas de maneras diferentes, a unas condiciones lo más cercanas a las del sistema fisiológico humano, para tener un parámetro de degradación, ya que la temperatura y el pH afectan directamente este proceso mediante la hidrólisis de estos poliésteres.
En la Figura 1 se muestran las micrografías de los cambios morfológicos.
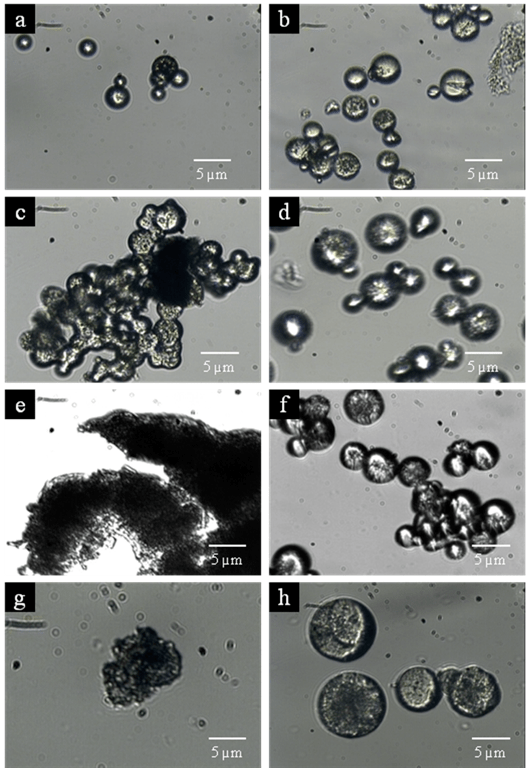
Se puede observar que a las primeras 7 horas de exposición en el medio, las microcápsulas aun conservaban su morfología, y algunas poseían pequeñas erosiones superficiales. A los 3 días de exposición (72 horas), la pérdida en la morfología de PDL fue evidente, mientras que PL aún mantuvo su morfología esférica. Posteriormente, al mes de exposición (30 días), se notó una disgregación de la masa amorfa en las microcápsulas de PDL, mientras que PL permaneció en su forma esférica con erosiones en su superficie.
Al transcurrir los 103 días de ensayo (casi 3 meses y medio), se mantuvo la masa amorfa de PDL (en menor cantidad), pero las microcápsulas de PL siguieron manteniendo su morfología esférica, con grandes erosiones en su superficie. La diferencia en la degradación fue significativa. Aun y cuando se trata de polilactida o poliácido láctico, la estereoquímica asociada al carbono metínico de la lactida o ácido láctico, confiere propiedades estructurales diferentes en cuanto al arreglo cristalino (como se verificó en la caracterización por 13C–RMN), dando como resultado un polímero amorfo de PDL y semicristalino de PL, lo cual conlleva a que estos materiales presenten diferencias en su degradación hidrolítica. En la bibliografía se ha reportado, que el polímero PDL es amorfo y el PL es semi–cristalino [9,10], y esto produce, en cuanto a la hidrólisis, que el PDL sea más fácil de degradar debido a que el ataque nucleofílico del agua sobre el carbono carbonílico del éster está más favorecido, mientras que PL por tener un arreglo semi–cristalino (zonas del polímero donde hay cadenas más ordenadas, ya que también hay zonas amorfas), presenta menos disponibilidad del carbono carbonílico del éster para que sea atacado nucleofílicamente por el agua, haciendo que sea más resistente a la hidrólisis, y que el ataque se dé principalmente en las zonas amorfas. En general, los materiales poliméricos con estructuras semi–cristalinas son más resistentes a la hidrólisis que los que tienen estructuras amorfas [10].
Caracterización por MEB de microcápsulas con contenido de CX generadas por el método de doble emulsión por evaporación de disolvente. La morfología externa de las microcápsulas de PL con CX, muestra una superficie suave con pequeños agujeros, son esféricas, y poseen una estructura interna tipo panal o reservorio, atribuida también a gotas de la solución de fármaco atrapadas dentro de las microcápsulas (Figura 2b). En el caso de PDL, son partículas esféricas, con superficie externa rugosa (Figura 2d). La estructura interna es un tanto diversa mostrando una morfología tipo cáscara (Figura 3d) y reservorio (específicamente en la Figura 3f donde se visualizan poros internos), con grandes agujeros a diferencia de la estructura interna de PL (Figuras 3a, 3b, y 3c). Esta diferencia en la estructura interna, se puede deber a una coalescencia de las gotas de la fase acuosa del fármaco de la primera emulsión en las microcápsulas de PDL, las cuales se concentraron en la matriz polimérica, en algunas microcápsulas, mientras que probablemente en la mayoría de ellas y en PL, la coalescencia de las gotas no fue completa y quedaron incrustadas en la matriz una vez que se evaporó el disolvente para formar las microesferas.


En las Figuras 4 y 5 se muestra la distribución de tamaño para una población de 67 microcápsulas de PDL con CX y 53 microcápsulas de PL con CX:
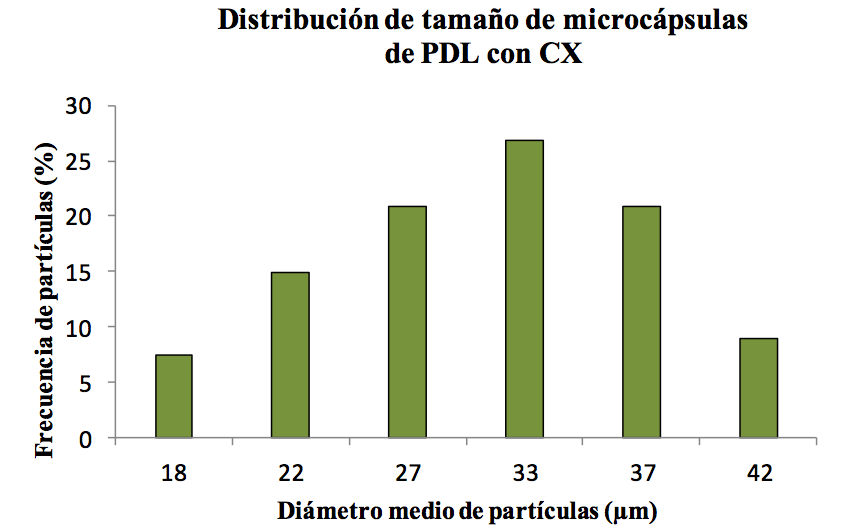
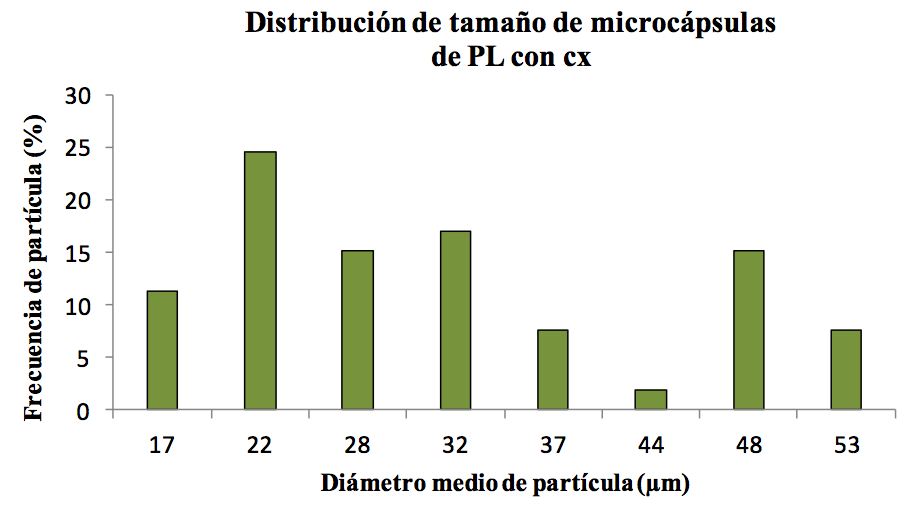
En la distribución de tamaño para las microcápsulas de PDL, se observa que la mayoría de las microcápsulas (69%) tienen diámetros medio que van de 27 ± 1 µm a 37 ± 1 µm, estadísticamente. El otro 31% se distribuye en diámetros medio de 22 ± 1 µm (15%), 42 ± 2 µm (9%) y 18 ± 2 µm (7%). En el caso de las microcápsulas de PL, un 25% de las microcápsulas posee un diámetro medio de 22 ± 1 µm, y el otro 75% se distribuye en tamaños entre 32 ± 2 µm (17%), 48 ± 1 µm (15%), 28 ± 2 (15%), 17 ± 4 (11%), 37 ± 1 µm (7,5%), 53 ± 1 µm (7,5%) y 44 (2%).
La polidispersidad e incremento de tamaño para ambos polímeros, se atribuye a factores de agitación que perturbaron la encapsulación homogénea y generación de partículas más pequeñas. No obstante, Jyotsana y col. [11] y Román y Chinea [12], emplearon condiciones semejantes para una microencapsulación con una doble emulsión, obteniendo una distribución de tamaño mono–dispersa de 17,8 µm (8.000 rpm) y 0–5 µm (750 rpm), respectivamente, lo cual también nos permite decir que es posible tener distribuciones de tamaño estrechas si se mantiene una agitación eficiente.
Eficiencia de encapsulación de CX en las microcápsulas. En la Tabla 1 se muestra un resumen de los valores obtenidos por espectroscopia UV–Visible usando la curva de calibración de CX en diclorometano:
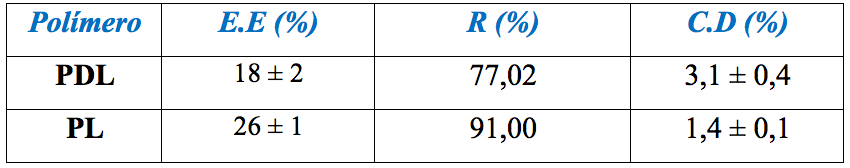
La eficiencia de encapsulación para ambas formulaciones se encuentra en los rangos reportados en la literatura, como en el caso de Román y Chinea cuya E.E fue 26,2% [12], Morgado y Chinea [13] 12,4% (para el homopolímero), y Jyotsana y col. [11] que reportaron eficiencias de encapsulación en un intervalo de 26–57%, de acuerdo a las condiciones empleadas para cada microencapsulación. Esto nos permite decir que los valores obtenidos son aceptables, ya que existen muchos factores en el proceso de microencapsulación que influyen en la eficiencia de encapsulación, tal como la solubilidad que posee el fármaco en la solución acuosa, que impide de entrada, la encapsulación completa debido a que este se difunde a través de la fase orgánica hacia la segunda fase acuosa, disminuyendo así la cantidad de fármaco disponible, en la primera emulsión, para encapsular.
El contenido de droga es bajo, pero también es un buen resultado debido a que hay que considerar lo riguroso de encapsular en pequeños microporos cierta cantidad de fármaco.
En cuanto al rendimiento de producción de cápsulas, se observa que en el proceso de producción de microcápsulas no hubo muchas pérdidas al momento de recolectar el producto con respecto a PL, mientras que en PDL hubo más pérdidas, posiblemente a microcápsulas adheridas a las paletas del agitador, que no fueron removidas eficientemente.
Perfil de liberación de CX por degradación de las microcápsulas. Ensayo in vitro en medio LIT. Para ambos perfiles, se observó una liberación de fármaco bifásica (Figura 6), lo cual implica una primera fase con un aumento de concentración de medicamento en el primer día para PDL, y PL. Este proceso se conoce como efecto de estallido inicial (burst, en idioma inglés) el cual se atribuye a la cantidad de fármaco que queda adherido a la superficie externa de la microcápsula y al que se encuentra internamente más cerca de la superficie, que es expulsado con mayor rapidez al entrar en contacto con un medio acuoso; una vez que se generan las primeras erosiones, comienza una estabilización en las curvas, segunda fase, donde la concentración de fármaco se libera de manera más controlada desde la matriz; en esta fase ya la expulsión del medicamento es por difusión a través de las porosidades de la microcápsula.
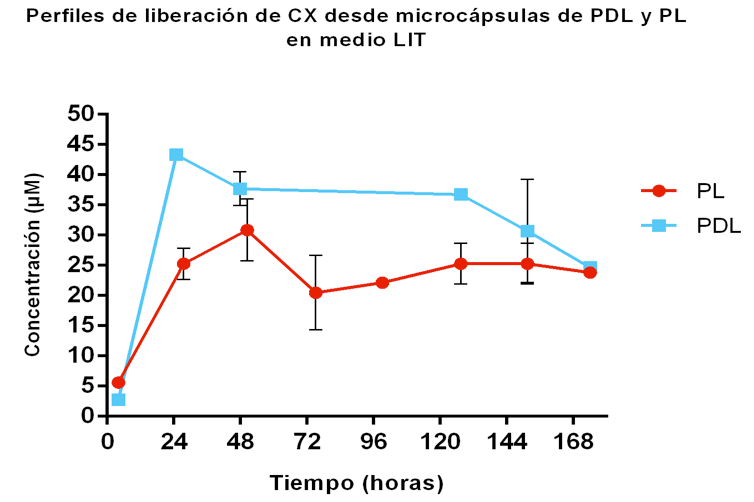
Se observó que, la liberación de CX los primeros días fue un tanto más controlada para las microcápsulas de PL mientras que en el caso de PDL, el efecto burst fue más significativo. La concentración alcanzada por los dispositivos de PL los dos primeros días fue 30 ± 5 µM, concentración usada como control en el ensayo de la curva de crecimiento, esto demuestra además que efectivamente el dispositivo es capaz de liberar una concentración deseada de acuerdo al sistema de trabajo (y a una cantidad de microcápsulas). En el caso de PDL la concentración de CX liberada durante los primeros 2 días alcanzó una liberación de CX de casi 43 ± 14 µM. La mayor concentración liberada por este dispositivo se adjudica también a que su degradación hidrolítica es mayor que la de PL.
Determinación de la viabilidad de promastigotes de Leishmania braziliensis en presencia de microcápsulas cargadas con CX mediante conteos directos. Curva de crecimiento. De acuerdo al perfil de liberación en medio LIT mostrado por las microcápsulas, se procedió a evaluar el efecto parasiticida del CX dispensado por las mismas sobre promastigotes de L. braziliensis. A continuación en la Figura 7 se muestra el resultado de los conteos realizados para este ensayo:
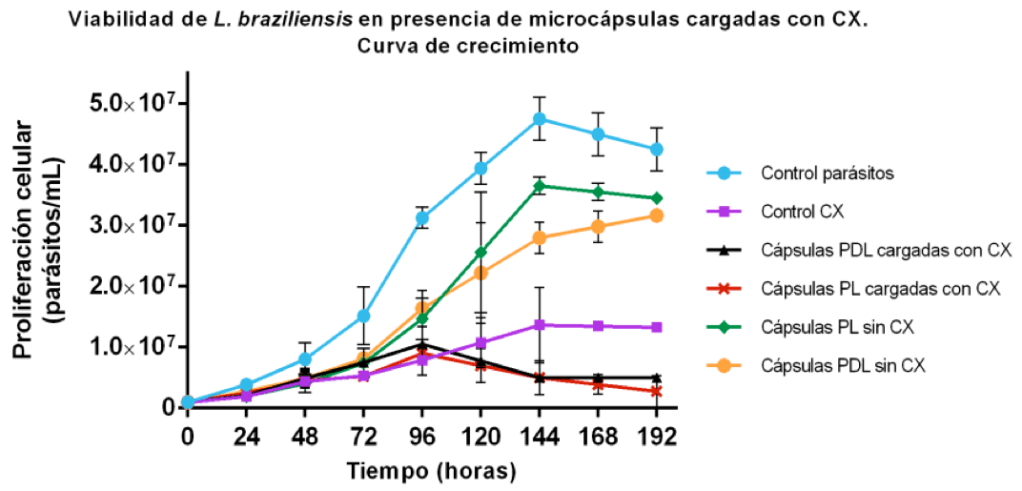
Se visualizó en la curva de control de parásitos (sin presión selectiva), un crecimiento en fase exponencial durante 7 días, lo cual en comparación con la curva de control de CX (sin encapsular) y de las microcápsulas con CX, muestra que los parásitos se encontraban en óptimas condiciones biológicas para replicarse naturalmente. Transcurrido este tiempo, observamos una fase estacionaria propia de sistemas con baja disponibilidad de nutrientes. Las curvas de control de CX y de microcápsulas con CX, mostraron un efecto parasiticida (momentáneo en el caso del control CX), lo cual ejerció una presión selectiva sobre los parásitos causando su declive durante las primeras etapas del ensayo. No obstante, en el control CX a los 6 días de experimento se observó un repunte moderado en la proliferación de los parásitos, en respuesta a una supervivencia de los mismos que los llevó a sobreponerse de la presión ejercida por el fármaco. Esto se aprecia justo a las 120 horas de ensayo donde los parásitos expuestos a CX libre proliferan, mientras que aquellos expuestos a microcápsulas con fármaco, disminuyen su proliferación de manera notable. Una posible justificación para esta diferencia en ambas condiciones es la siguiente: el crecimiento de los parásitos que ocurre después de un tiempo determinado con la dosis de fármaco directa, puede deberse a un consumo del medicamento en presencia de los parásitos, aunque no se siguieron los cambios de concentración de CX a través de espectrofotometría UV–Visible, se podría justificar este consumo debido a la inhibición de enzimas en microorganismos que poseen cofactores metálicos tales como Fe+3 que son quelados por el ciclopirox. Se ha reportado que este fármaco posee características de agente quelante [14]. En la literatura se demostró que el ciclopirox inhibió una enzima llamada DOHH (desoxihipusina hidroxilasa) sobre parásitos L. donovani, la cual culmina la biosíntesis de hipusina (un aminoácido presente en las células eucariotas). El mecanismo de acción que justificó este efecto, fue considerando al CX como agente quelante, ya que esta enzima posee un metal de Fe2+ en su centro [15]. Aunado a ello también se ha alegado que el hierro es vital para todos los parásitos trypanosomatideos (lo cual incluye el género Leishmania spp) y juega un papel fundamental en la patogénesis y el control inmunitario de estos organismos. En este sentido, el agotamiento de este nutriente esencial en los trypanosomatideos disminuye rápidamente la tasa de síntesis de ADN, impidiendo la formación de sustancias vitales en el protozoario, lo que al final lleva consigo la muerte de los mismos [16]. Debido a esta justificación, se infiere que añadir una concentración de CX, en este caso de 30 µM, afecta la proliferación parasitaria durante un período temporal, donde luego este efecto se ve disminuido, ya que progresivamente se va consumiendo el medicamento y esto genera más bien un efecto parasitostático, es decir, se inhibe la rápida proliferación de parásitos, logrando así que estos se recuperen lentamente de la presión selectiva. Ahora, en el caso de las microcápsulas con fármaco, se nota un significativo efecto parasiticida sostenido a partir de las 96 horas de cultivo. Al parecer, la liberación sostenida del fármaco, afecta de manera constante la viabilidad de los parásitos que se van replicando por lo que estos no pueden responder ante la presión selectiva. Pareciera entonces que en términos terapéuticos, aplicar un dispositivo de liberación controlada de CX, es más efectivo que añadir una única dosificación del fármaco libre.
En cuanto a las microcápsulas sin fármaco, se notó un efecto ligeramente parasiticida. Este mismo efecto fue reportado en el trabajo de Pesquera et al. [5]con copolímerosdepoli(lactida–co–glicolida), donde infirieron que este efecto se debe a microclimas ácidos o µpH ácidos, localizados en los poros que se generan en las cápsulas, donde están disueltas las especies oligoméricas provenientes de la degradación del polímero [17], que posiblemente produjo la baja proliferación parasitaria, sin embargo, al medir el pH de los medios en estudio con papel tornasol, no registraron un descenso del pH. Teniendo en consideración esta hipótesis, se podría inferir que una pequeña población de parásitos quedó inmersa en estos poros con estos microclimas ácidos, lo que afectó la viabilidad de los mismos. No obstante, sería pertinente realizar ensayos más específicos que permitan justificar este resultado.
CONCLUSIONES
La síntesis por ROP permitió obtener homopolímeros de poli (D,L y L) lactida, con arreglos cristalinos y tacticidades diferentes de acuerdo a la estereoquímica asociada al carbono metínico de los monómeros de lactida, que confirió una estructura y comportamiento diferente de ambos polímeros en los ensayos in vitro. También fue posible encapsular el CX con el método de doble emulsión por evaporación de solvente, logrando que los dispositivos lo dispensaran de manera controlada sobre ensayos en medios acuosos a una temperatura determinada. Las microcápsulas lograron dispensar de manera controlada el CX sobre L. braziliensis, mostrando un efecto parasiticida efectivo y sostenido con respecto al CX añadido directamente en donde los parásitos fueron capaces de sobreponerse a la presión selectiva.
Los resultados mostrados, resultan alentadores en la búsqueda de nuevas estrategias para el futuro tratamiento de pacientes con leishmaniasis cutánea, sin embargo, son necesarios más investigaciones orientadas al ajuste de concentraciones letales, empleo y dosificación en mamíferos. Ensayos in vivo (ratones experimentales), están siendo desarrollados actualmente por nuestro grupo de trabajo.
Agradecimientos. Los autores reconocen y aprecian la contribución de los investigadores de los Centros de Catálisis, Petróleo y Petroquímica, Equilibrio en Solución, Microscopía Electrónica “Mitsuo Ogura” y Fisicoquímica de la Universidad Central de Venezuela), así como los del Laboratorio de Biología y Quimioterapia de Parasitosis Tropicales, Áreas de Salud, Agricultura y Soberanía Alimentaria de la Fundación Instituto de Estudios Avanzados (IDEA). También se hace extensivo el agradecimiento a CDCH por financiar el proyecto 03PG7821, para Carlos Chinea y FONACIT por financiar los proyectos 2012002132, 2012000784 y 201201961 para Xenón Serrano–Martín.
BIBLIOGRAFÍA
[1] Chasin, M., Langer, R. (1990) Biodegradable polymers as drug delivery systems. Marcel Dekker, New York
[2] Lopes, M. S., Jardini, A. L., Filho, R. M. (2014). Synthesis and characterizations of poly (lactic acid) by ring-opening polymerization for biomedical applications. Chemical Engineering Transactions, 38, 331-336.
[3] Mehta, R., Kumar, V., Bhunia, H., & Upadhyay, S. N. (2005). Synthesis of poly (lactic acid): a review. Journal of Macromolecular Science, Part C: Polymer Reviews, 45(4), 325-349.
[4] Msuya, N., Katima, J. H. Y., Massanja, E., & Temu, A. K. (2017). Poly (lactic-acid) Production-from Monomer to Polymer: A Review. SciFed Journal of Polymer Science, 1(1).
[5] Pesquera, L. (2002). Síntesis y caracterización de microcápsulas de ácido poli–(láctico–co–glicólico). Evaluación de sus propiedades como agente de liberación controlada en la quimioterapia contra la leishmaniasis (Tesis de grado, Universidad Central de Venezuela, Caracas, Venezuela).
[6] Vysloužil, J., Doležel, P., Kejdušová, M., Mašková, E., Mašek, J., Lukáč, R., Košťál, V., Vetchý, D. & Dvořáčková, K. (2014). Influence of different formulations and process parameters during the preparation of drug-loaded PLGA microspheres evaluated by multivariate data analysis. Acta Pharmaceutica, 64(4), 403-417.
[7] Bero, M., Kasperczyk, J., & Jedlinski, Z. J. (1990). Coordination polymerization of lactides, 1. Structure determination of obtained polymers. Die Makromolekulare Chemie: Macromolecular Chemistry and Physics, 191(10), 2287-2296.
[8] Azevedo H. S., Reis R. L. (2004) Understanding the enzymatic degradation of biodegradable polymers and strategies to control their degradation rate in Biodegradable systems in tissue engineering and regenerative medicine (Reis, R. L., & San Román, J. eds.). CRC press.
[9] Tsuji, H., & Ikada, Y. (1997). Blends of crystalline and amorphous poly (lactide). III. Hydrolysis of solution‐cast blend films. Journal of applied polymer science, 63(7), 855-863.Chicago
[10] Hyon, S. H., Jamshidi, K., & Ikada, Y. (1998). Effects of residual monomer on the degradation of DL‐lactide polymer. Polymer international, 46(3), 196-202.
[11] Madan, J., Kadam, V., Bandavane, S., & Dua, K. (2013). Formulation and evaluation of microspheres containing ropinirole hydrochloride using biodegradable polymers. Asian Journal of Pharmaceutics, 7(4), 184-188.
[12] Román, M. (2005). Estudio de la velocidad de liberación de sustancias encapsuladas y caracterización morfológica de sistemas poliméricos biodegradables utilizando técnicas de UV y fluorescencia láser confocal (Tesis de grado, Universidad Central de Venezuela, Caracas, Venezuela).
[13] Morgado, M. (2006). Estudio de la inhibición de Monoamino Oxidasa (MAO) por deprenilo mediante uso de polímeros biodegradables como agente de liberación controlada (Tesis de grado, Universidad Central de Venezuela, Caracas, Venezuela).
[14] Subissi, A., Monti, D., Togni, G., & Mailland, F. (2010). Ciclopirox. Drugs, 70(16), 2133-2152.
[15] Chawla, B., Kumar, R. R., Tyagi, N., Subramanian, G., Srinivasan, N., Park, M. H., & Madhubala, R. (2012). A unique modification of the eukaryotic initiation factor 5A shows the presence of the complete hypusine pathway in Leishmania donovani. PloS one, 7(3), e33138.
[16] LS Santos, A., L Sodre, C., S Valle, R., A Silva, B., A Abi-chacra, E., V Silva, L., L Souza-Goncalves, A., S Sangenito, L., S Goncalves, D., OP Souza, L. and F Palmeira, V. (2012). Antimicrobial action of chelating agents: repercussions on the microorganism development, virulence and pathogenesis. Current medicinal chemistry, 19(17), 2715-2737.
[17] Ding, A. G., Shenderova, A., & Schwendeman, S. P. (2006). Prediction of microclimate pH in poly (lactic-co-glycolic acid) films. Journal of the American Chemical Society, 128(16), 5384-5390.
