Luis Pesquera1,2, Xenón Serrano–Martín2,3,Jorge Núñez2, Carlos Chinea1*
1) Laboratorio de Polímeros, Escuela de Química, Universidad Central de Venezuela (UCV). Caracas, Venezuela. Correo electrónico: carlos.chinea@ciens.ucv.ve
2) Laboratorio de Biología y Quimioterapia de Parásitos Tropicales (ByQPAT), Instituto de Estudios Avanzados (IDEA). Caracas, Venezuela. Correo electrónico: xenonserrano@gmail.com
3) Centro de Química Orgánica, Facultad de Ciencias, Universidad Central de Venezuela (UCV). Caracas,Venezuela.
Recibido: Julio 2019; Aceptado: Agosto 2019
Texto completo (pdf)
Cita (APA)
Pesquera, L., Serrano-Martín, X., Núñez, J., Chinea, C. (2019). Síntesis y caracterización de microcápsulas de ácido poli–(láctico–co–glicólico). Evaluación de sus propiedades como potencial agente de liberación controlada, en la quimioterapia contra la leishmaniasis cutánea. Revista Iberoamericana de Polímeros, 20(5), 196–206.
RESUMEN
La leishmaniasis cutánea es una enfermedad parasitaria tropical caracterizada por el surgimiento de lesiones dérmicas como foco de la infección, reconocida como un problema de salud pública en varios países. En este trabajo se realizó el estudio del efecto de la composición de monómeros en un polímero lactida–co–glicolida, en la cinética de liberación de ciclopirox olamina (Cx) y el estudio de la citotoxicidad de Cx en macrófagos. La síntesis del poli(láctida–co–glicólida) (PLGA), se realizó por reacción de apertura de anillo (ROP). Las microcápsulas fueron preparadas utilizando el método de doble emulsión–evaporación, donde se obtuvieron partículas con una superficie lisa y morfología esférica. Las cápsulas del PLGA 95:5 comenzaron a degradarse a partir de los 12 días, mientras que a los 40 días; ambos dispositivos sufren coalescencia y descomposición extensivas. Finalmente, la evaluación biológica mostró una notable afectación de la viabilidad parasitaria, a concentraciones inferiores a 20 μM de Cx. Logramos demostrar que Cx afecta levemente la viabilidad de macrófagos BMDM a concentraciones por encima de 20 μM, lo que se traduce en una evidente toxicidad selectiva hacia el parásito. Los resultados, resultan alentadores en la búsqueda de nuevas estrategias para el potencial tratamiento de pacientes con Leishmaniasis Cutánea.
ABSTRACT
Cutaneous leishmaniasis is a tropical parasitic disease characterized by the appearance of dermal lesions as the focus of infection, recognized as a public health problem in several countries. In this work, the study of the effect of monomer composition in a lactide–co–glycolide polymer, in the kinetics of ciclopirox olamine (Cx) release and the study of Cx cytotoxicity in macrophages was carried out. The synthesis of poly (lactic acid–co–glycolide) (PLGA) was performed by ring opening reaction (ROP). The microcapsules were prepared using the double emulsion–evaporation method, where particles with a smooth surface and spherical morphology were obtained. The PLGA 95:5 capsules began to degrade after 12 days, while at 40 days; both devices suffer extensive coalescence and decomposition. Finally, biological evaluation showed a significant affectation of the parasitic viability, at concentrations lower than 20 μM of Cx. We were able to demonstrate that Cx slightly affects the viability of BMDM macrophages at concentrations above 20 μM, which translates into an evident selective toxicity towards the parasite. The results are encouraging in search for new strategies for the potential treatment of patients with Cutaneous Leishmaniasis.
INTRODUCCIÓN
La leishmaniasis es una enfermedad endémica de zonas tropicales que surge como consecuencia de infecciones causadas por varias especies de parásitos protozoarios del género Leishmania spp. De acuerdo a la Organización Mundial de la Salud (OMS), la leishmaniasis figura entre las seis parasitosis con los mayores índices de mortalidad y morbilidad en el planeta [1]. En el caso particular de Venezuela, alrededor del 98% de los casos presenta síntomas de la variable cutánea de la enfermedad (forma con mayor índice de morbilidad, pero con mucho menores índices de mortalidad que la variable visceral), y de esos casos, el 80% es causado por una única especie de parásito: Leishmania braziliensis [2].
Los parásitos del género Leishmania spp presentan dos morfotipos bien establecidos a lo largo de su ciclo de vida, observables según el hospedador en el que el parásito se encuentre (el ciclo de vida de éste es digénico, pues se alterna entre dos hospedadores, uno vertebrado y otro invertebrado). El primer morfotipo es conocido como promastigote, forma flagelada del parásito cuyo desarrollo se efectúa en el sistema digestivo del hospedador invertebrado (en el ciclo de vida del parásito, estos hospedadores participan como vectores). El segundo morfotipo se denomina amastigote, y se genera como consecuencia de una transformación que sufre la forma promastigote al ser fagocitada por los macrófagos (un tipo de célula perteneciente al sistema inmunológico) del hospedador vertebrado [2].
La disponibilidad actual de fármacos para el tratamiento es baja y la mayoría de ellos se administra por vía parenteral, la especificidad de acción terapéutica y toxicidad aumenta la posibilidad de efectos adversos, así como el surgimiento de fenómenos de resistencia por parte de los parásitos causantes de la enfermedad [2].
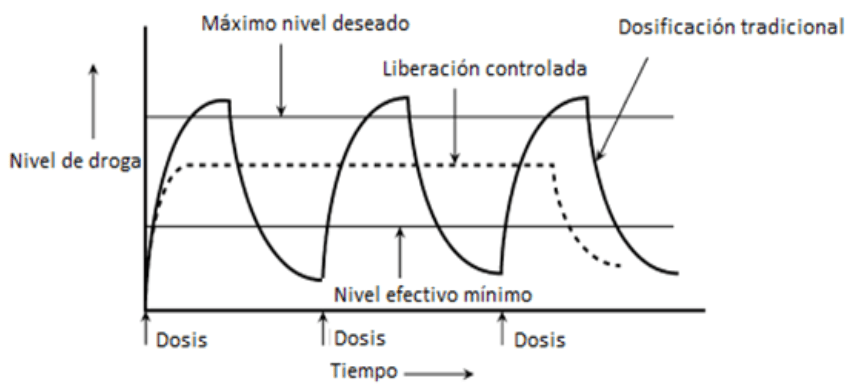
Los polímeros bio–compatibles y biodegradables, los cuales, no generan respuestas inmunológicas en el paciente [3] y el hecho de que sus productos de degradación puedan ser metabolizados por vías fisiológicas normales del organismo [4], representan una alternativa para suplantar lo métodos de administración convencionales. El objetivo de cualquier sistema de liberación controlada de fármacos dosificar la sustancia en un lugar y tiempo específicos, bajo un patrón de liberación; a diferencia de los métodos clásicos de administración de medicamentos (píldoras, inyecciones), que comprenden liberaciones en forma de “picos”, los cuales alcanzan concentraciones por encima de la dosis requerida (véase la Figura 1 [3]).
El co–polímero ácido poli(láctico–co–glicólico) (PLGA), permite variar la liberación del fármaco encapsulado desde semanas hasta meses [4], dependiendo de factores como su peso molecular, cristalinidad, tacticidad, proporciones de monómeros. El método de síntesis más frecuentemente empleado para su obtención es la polimerización por apertura de anillo (ROP, por sus siglas en inglés, Figura 2) de dímeros cíclicos de ácido láctico y ácido glicólico, denominados lactida y glicolida respectivamente, de acuerdo a la siguiente reacción general [5].

El PLGA comienza a ser sintetizado a partir de inicios de la década de 1970, con la patente registrada por Boswell y Scribneren en 1973 [6] donde, por primera vez, se realizan preparaciones con diversos sistemas droga/polímero.
El objetivo de este trabajo es realizar la síntesis del PLGA en dos relaciones másicas diferentes, la encapsulación de Cx [7] en la matriz polimérica, evaluación de las características físicas (distribución de tamaño, morfología, tiempo de descomposición de la matriz polimérica) y el comportamiento de las cápsulas como agente de liberación (cinética de liberación del fármaco en medio fisiológico). Además, pretendemos estudiar la acción “in vitro”, del fármaco liberado sobre promastigotes de Leishmania braziliensis y sobre macrófagos BMDM obtenidos de ratones de experimentación.
PARTE EXPERIMENTAL
Síntesis del PLGA vía ROP. En ampollas de vidrio previamente secadas y en atmósfera inerte, se añadieron 10 g de los monómeros, 99:1 a 95:5 de proporciones de D,L lactida y glicolida, respectivamente. Luego, se agregaron 5 mL de una suspensión de cloruro de estaño (II) dihidratado en dietiléter, y otros 5 mL de una solución de 1–dodecanol en dietiléter. Todas las especies mencionadas fueron mezcladas mediante agitación magnética por una hora, para luego evaporar el solvente a través de vacío. Finalmente, las ampollas fueron selladas y se sumergieron en baño térmico a 160°C durante 8 horas [8–10]. Finalizadas las 8 horas de síntesis, los reactores se almacenaron en el congelador para detener la reacción. El producto fue disuelto en una mínima cantidad de cloroformo, precipitándose seguidamente en metanol. El polímero final fue filtrado por gravedad, secado al vacío y almacenado en atmósfera inerte y ambiente refrigerado.
Elaboración de las microcápsulas. Las microcápsulas fueron preparadas usando el método de doble emulsión (w/o/w) con evaporación de solvente. La primera emulsión w/o, se preparó a partir una solución al 0,05% en masa de polímero en 10 mL de cloroformo y una fase acuosa, donde se disolvieron 7,4 mg de fármaco Cx en 1,6 mL de solución acuosa de agar al 1,8% en masa a 60°C. Las dos soluciones fueron mezcladas, gota a gota, en un baño de agitación con ultrasonido. Para preparar la segunda emulsión w/o/w, se agregó la primera emulsión en 200 mL de una solución acuosa de polivinil alcohol (PVOH) al 2% en agitación a una velocidad de 800 rpm y mantenida a 15°C durante 30 minutos. Al transcurrir este tiempo, se redujo la velocidad de agitación a 500 rpm, se eliminó el baño frio y se continuó el proceso de agitación por un lapso adicional de 5 horas. La suspensión obtenida fue centrifugada, y las microcápsulas fueron lavadas con agua destilada, secadas al vacío, pesadas y almacenadas en atmósfera inerte y ambiente refrigerado [8–10].
Evaluación del tiempo de descomposición de las microcápsulas en condiciones fisiológicas emuladas. Aproximadamente 5 mg de microcápsulas, preparadas sin fármaco en su interior, fueron colocadas en tubos de ensayo que contenían, 1 mL de buffer de fosfato (PBS, pH = 7). Los tubos fueron parcialmente sumergidos en un baño de agua a una temperatura constante de 37°C por un periodo de tiempo que permitiera conseguir la degradación total del material. Para evaluar el estado morfológico de las microcápsulas, se tomaron alícuotas de suspensión de microcápsulas cada cierto tiempo, y las mismas fueron observadas al microscopio óptico.
Determinación de la cantidad de fármaco encapsulado. Se prepararon soluciones madre de cada de copolímero disolviendo 1 mg de microcápsulas con fármaco en 10 mL de cloroformo. A partir de cada solución madre, se prepararon soluciones más diluidas para hacer determinaciones cuantitativas de concentración del fármaco contenido en las cápsulas mediante espectrometría UV–Visible (longitud de onda de trabajo: 306 nm). Las concentraciones fueron determinadas utilizando una curva de calibración de concentración del fármaco en cloroformo.
Cinética de liberación del fármaco desde las cápsulas. En una placa microtituladora de 24 pozos, se dispusieron 2 mL de solución de buffer de fosfato en dos pozos diferenciados. En cada uno de estos pozos, se coloca entre 0,6–0,7 mg de cápsulas de polímero con fármaco en su interior. La placa se dejó reposar en un equipo con agitación y control de temperatura (28–30°C) durante 30 días, tomándose alícuotas de 200 μL y sometiéndolas a medidas espectrofotométricas UV–Visible a 306 nm y devolviendo tales alícuotas a su placa original. A partir de una curva de calibración previamente construida, la concentración del fármaco liberado para cada lapso de tiempo pudo obtenerse. Al seleccionar el dispositivo que presentó mejor desempeño, su cinética de liberación en medio LIT (medio de cultivo de los parásitos de Leishmania braziliensis) también fue determinada del mismo modo ya descrito.
Viabilidad de promastigotes de Leishmania braziliensis en presencia de microcápsulas con ciclopirox olamina. Se dispusieron, en varias fiolas, 10 mL de medio LIT con una concentración de 1·106 parásitos por mililitro, y a cada triplicado de fiolas se le agregó, bajo condiciones de estricta esterilidad, lo siguiente: 1) Primer triplicado: 0,5 mg de microcápsulas por fiola. 2) Segundo triplicado: cápsulas vacías. 3) Tercer triplicado: solución de Cx no encapsulada a una concentración 150 μM. 4) Cuarto triplicado: parásitos sin cápsulas ni fármaco. Las fiolas se mantuvieron en agitación permanente y a temperatura de 29°C, tomándose alícuotas de todas las condiciones cada 24 horas para efectuar conteos directos de proliferación parasitaria en cámara de Neubauer por un total de 11 días. A partir de los conteos directos, se empleó la siguiente ecuación para determinar la población parasitaria.
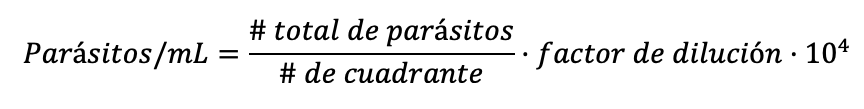
Estudio de la citotoxicidad del ciclopirox olamina en macrófagos. Macrófagos derivados de médula ósea de ratón (macrófagos BMDM) fueron depositados sobre placas cubreobjetos (80 μL de medio de cultivo DMEM con 8·103 macrófagos por placa), las cuales, a su vez, fueron dispuestas dentro de placas microtituladoras de 6 pozos. Las placas fueron incubadas a 37°C, 5% CO2 durante 24 horas, tras lo cual se sometieron los macrófagos a diversas condiciones (Figura 3): 1) Primer triplicado: 100 μM Cx. 2) Segundo triplicado: 300 μM Cx.3) Tercer triplicado: 500 μM Cx. 4) Cuarto triplicado: 1.000 μM Cx. 5) Quinto triplicado: 100 μM de miltefosina (Control Mf, droga de uso comercial contra Leishmaniasis), 6) Sexto triplicado: macrófagos y cápsulas vacías, 7) Séptimo triplicado: macrófagos sin cápsulas ni fármaco (Control Mac), y 8) Octavo triplicado: medio de cultivo DMEM sin células.
La placa fue incubada a 37°C, 5% CO2, durante 96 horas. Seguidamente, se retiraron las placas cubreobjetos según se fuesen a contabilizar al microscopio, para lo cual se les adicionaron 10 μL de solución colorante de azul de tripano al 1%. Las células vivas contabilizadas fueron aquellas en la que no se internalizó el colorante, producto de las óptimas condiciones de su membrana celular.

RESULTADOS Y DISCUSIÓN
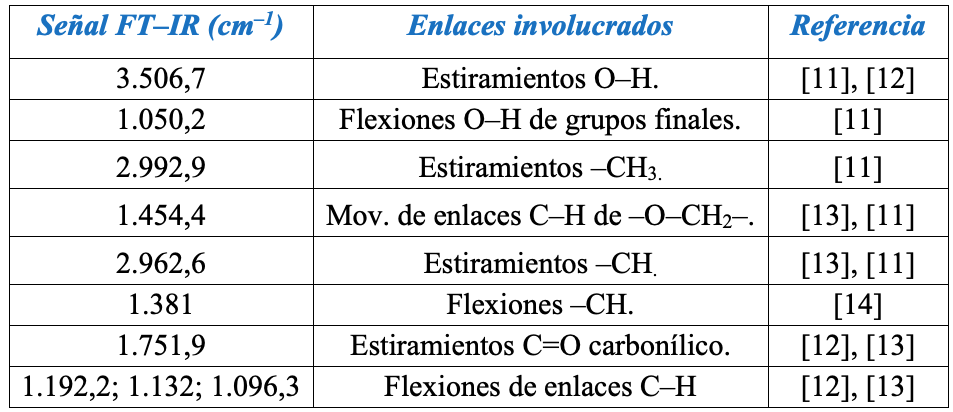
Síntesis del PLGA vía ROP. Los dos PLGA sintetizados con diferentes proporciones monoméricas exhibieron señales infrarrojas características de un α–hidroxi–poliéster, coincidiendo todos con la información de la Tabla 1.
En la Tabla 2, se muestran los desplazamientos químicos determinados a partir del espectro 1H–RMN para el PLGA 95:5.
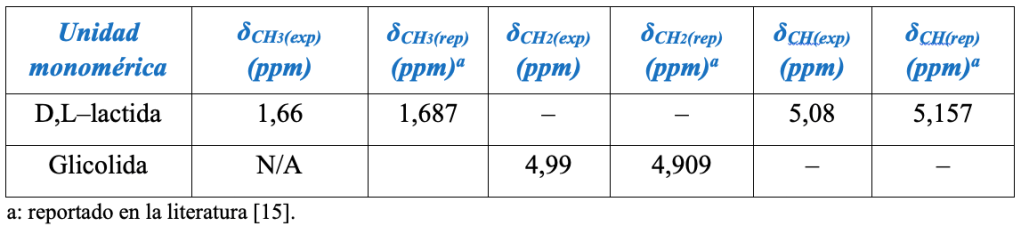
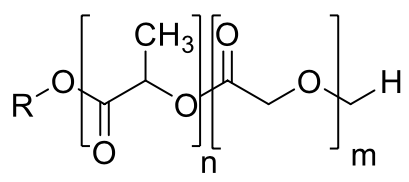
A partir de las integrales de las áreas de las señales y de las ecuaciones 2 y 3 [11] se pudo obtener la composición real de los PLGA, la cual, de acuerdo a lo mostrado en la Tabla 3, se correlaciona bien con la composición nominal esperada.


ELABORACIÓN Y CARACTERIZACIÓN DE LAS CÁPSULAS
Caracterización morfológica. En la figura 4 se muestran las micrografías obtenidas por microscopía electrónica de barrido (MEB)
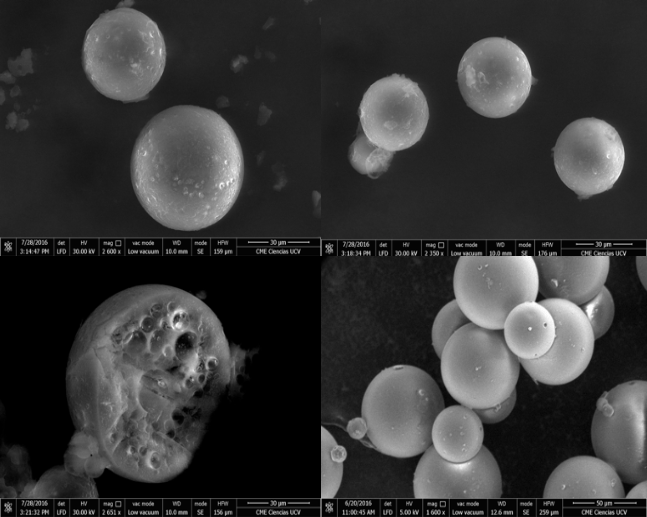
Se observa que las cápsulas poseen una superficie lisa y morfología esférica. Internamente, se observaron múltiples porosidades, posiblemente debido al pequeño tamaño de las gotas de la primera emulsión con respecto a la microcápsula final [12]. Una vez obtenidas las microcápsulas, se utilizó el software ImageJ para procesar micrografías y obtener una curva de distribución de tamaño (Figura 5).
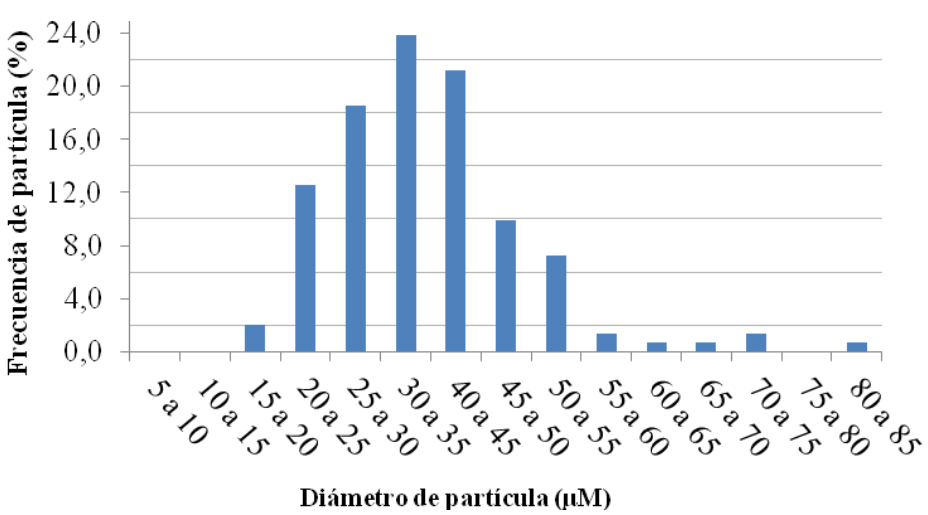
Esta curva permite conocer que más del 90% de la frecuencia de partícula se ubica entre 20 y 55 μm, con más del 60% acotada entre 25 y 45 μm.
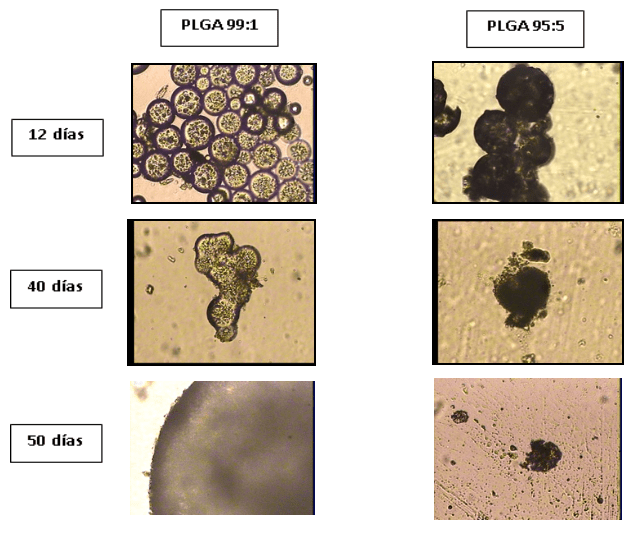
Tiempo de descomposición de las microcápsulas en condiciones fisiológicas emuladas. La evolución de la descomposición de las cápsulas se muestra en la Figura 6.
Cinética de liberación del fármaco desde las cápsulas. El gráfico cinético de liberación de fármaco a partir de las cápsulas se muestra a continuación, Figura 7.
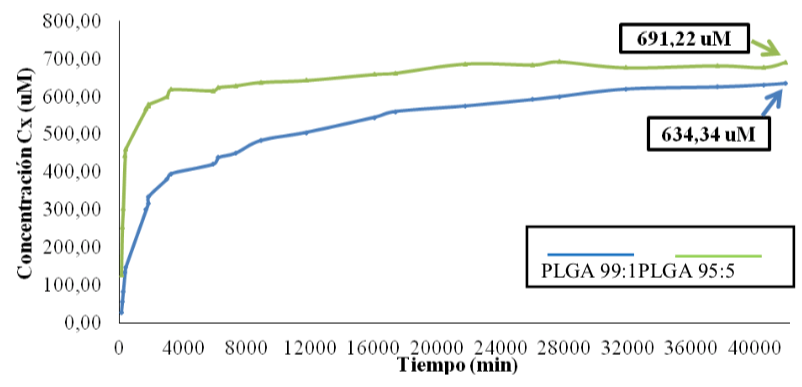
El perfil general revela, para ambos tipos de cápsulas, un patrón de liberación bimodal, con un efecto burst o de “explosión” inicial, normalmente adjudicado a la rápida expulsión de la droga asociada a la superficie interna de la matriz, y una posterior estabilización del régimen de liberación como segunda etapa del patrón bimodal. Regímenes bimodales muy similares se consiguieron también en medio LIT, aunque con mayores concentraciones liberadas, lo cual se adjudicó a un desgaste de las cápsulas por el tiempo de uso y manipulación en este punto de la investigación. Debido a que desde las cápsulas de PLGA 99:5 se dosificó el fármaco de manera mucho menos prolongada (casi 90% de su contenido total durante los primeros tres días, contra un 60% desde el sistema PLGA 99:1), el dispositivo PLGA 99:1 fue seleccionado para ser utilizado en los ensayos subsiguientes.
Viabilidad de promastigotes de Leishmania braziliensis en presencia de microcápsulas con ciclopirox olamina. La curva de crecimiento resultante se muestra en la Figura 8.
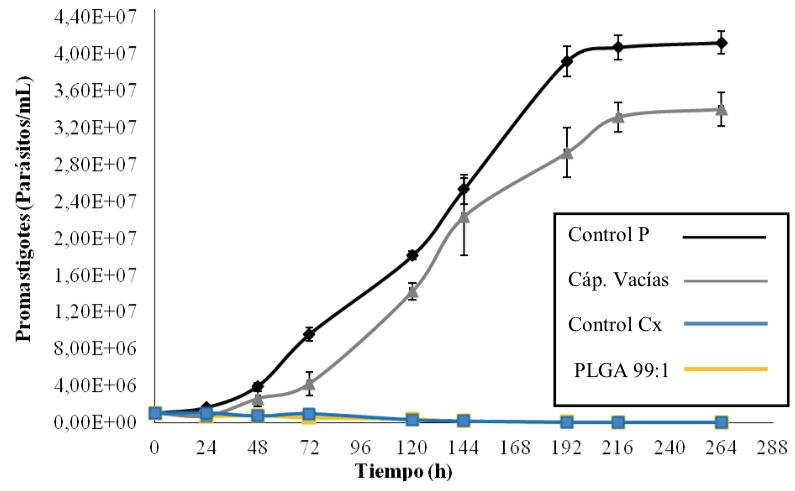
Los parásitos sin fármaco, ni cápsulas (Control P) exhibieron crecimiento exponencial los primeros 8 días, mientras que, tanto para el control con fármaco libre (Control Cx) como para el fármaco liberado desde las cápsulas, el desarrollo y crecimiento fueron inhibidos desde el inicio del experimento. Lo anterior, da cuenta de un evidente efecto antiparasitario. Notablemente, las cápsulas vacías también produjeron un efecto antiparasitario moderado, el cual pudiese potenciar, al generado por el Cx una vez se ingresa a la cápsula (ensayo en ejecución).
Estudio de la citotoxicidad del ciclopirox olamina en macrófagos. En la Figura 9, se representa la respuesta de las poblaciones de macrófagos a diversas condiciones ensayadas.
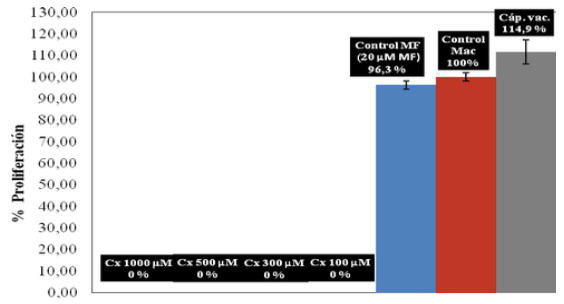
Los macrófagos expuestos a concentraciones por encima de 100 μM, mostraron una afectación total de su viabilidad celular. Sin embargo, cuando estos son expuestos a una concentración de 20 μM de Cx, observamos más de un 95% de sobrevida. Este dato demuestra en parte, la inocuidad del fármaco sobre las células hospederas y valida la acción específica sobre el parásito. Aunque este representa un ensayo preliminar, la evidencia representa un indicio interesante para nuevas investigaciones que verifiquen a profundidad lo observado.
CONCLUSIONES
Los métodos experimentales aplicados lograron sintetizar exitosamente el PLGA en las proporciones deseadas, y producir microcápsulas con fármaco encapsulado que, efectivamente, fungieron como agentes de liberación prolongada del mismo, variando su desempeño en función a su composición. El Cx, tanto libre como liberado, registró visible efecto tanto sobre la viabilidad de Leishmania braziliensis, afectando levemente la viabilidad de células hospederas (macrófagos BMDM). Los resultados mostrados, resultan alentadores en la búsqueda de nuevas estrategias para el tratamiento de pacientes con leishmaniasis cutánea, sin embargo, son necesarias más investigaciones orientadas al ajuste de concentraciones letales, empleo y dosificación en mamíferos.
Agradecimientos. Los autores reconocen y aprecian la contribución de los investigadores de los Centros de Catálisis, Petróleo y Petroquímica, Equilibrio en Solución, Microscopía Electrónica “Mitsuo Ogura” y Fisicoquímica de la Universidad Central de Venezuela, así como los del Laboratorio de Biología y Quimioterapia de Parasitosis Tropicales, Áreas de Salud y Agricultura y Soberanía Alimentaria de la Fundación Instituto de Estudios Avanzados (IDEA). También se hace extensivo el agradecimiento a CDCH por financiar el proyecto 03PG7821, para Carlos Chinea y FONACIT por financiar los proyectos 2012002132, 2012000784 y 201201961 para Xenón Serrano–Martín.
BIBLIOGRAFÍA
[1] Núñez J et al., Rev. Fac. Farm. UCV,75(2), 50 (2012) PENDIENTE
[2] Serrano-Martín, X. (2010). Quimioterapia contra la leishmaniasis: estado del arte, retos y nuevas propuestas desde Venezuela. RET. Revista de Estudios Transdisciplinarios, 2(1), 69-75.
[3] Stamatialis, D. F., Papenburg, B. J., Girones, M., Saiful, S., Bettahalli, S. N., Schmitmeier, S., & Wessling, M. (2008). Medical applications of membranes: drug delivery, artificial organs and tissue engineering. Journal of Membrane Science, 308(1-2), 1-34.
[4] Youxin, L., & Kissel, T. (1993). Synthesis and properties of biodegradable ABA triblock copolymers consisting of poly (L-lactic acid) or poly (L-lactic-co-glycolic acid) A-blocks attached to central poly (oxyethylene) B-blocks. Journal of controlled release, 27(3), 247-257.
[5] Babiuch, K., Gottschaldt, M., Werz, O., & Schubert, U. S. (2012). Particulate transepithelial drug carriers: barriers and functional polymers. RSC Advances, 2(28), 10427-10465.
[6] Boswell, G., & Scribner, R. (1973). U.S. Patent No. 3,773,919. Washington, DC: U.S. Patent and Trademark Office.
[7] Chawla, B., Kumar, R. R., Tyagi, N., Subramanian, G., Srinivasan, N., Park, M. H., & Madhubala, R. (2012). A unique modification of the eukaryotic initiation factor 5A shows the presence of the complete hypusine pathway in Leishmania donovani. PloS one, 7(3), e33138.
[8] Tortolero, M. (2002). Empleo de polímeros biodegradables sintéticos como sistemas de liberación controlada de drogas en el tratamiento de infecciones óseas (Tesis doctoral, Universidad Central de Venezuela, Caracas, Venezuela).
[9] Román, M. (2005). Estudio de la velocidad de liberación de sustancias encapsuladas y caracterización morfológica de sistemas poliméricos biodegradables utilizando técnicas de UV y fluorescencia láser confocal (Tesis de grado, Universidad Central de Venezuela, Caracas, Venezuela).
[10] Morgado, M. (2006). Estudio de la inhibición de Monoamino Oxidasa (MAO) por deprenilo mediante uso de polímeros biodegradables como agente de liberación controlada (Tesis de grado, Universidad Central de Venezuela, Caracas, Venezuela).
[11] D’Avila Carvalho Erbetta, C., Alves, R. J., Resende, J. M., de Souza Freitas, R. F., & de Sousa, R. G. (2012). Synthesis and characterization of poly (D, L-lactide-co-glycolide) copolymer. Journal of Biomaterials and Nanobiotechnology, 3(02), 208.
[12] Rosca, I. D., Watari, F., & Uo, M. (2004). Microparticle formation and its mechanism in single and double emulsion solvent evaporation. Journal of controlled release, 99(2), 271-280.
